Multiscale quantum models for ribozyme catalysis
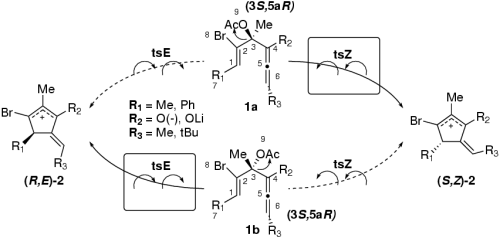
This project focuses on providing deeper insight into the molecular mechanisms of RNA catalysis through the development and application of state-of-the-art theoretical methods. Predictive modeling of RNA-catalyzed reactions needs to take into account accurate quantum models, complex macromolecular, ionic and solvent environments, and extensive conformational sampling. These requirements necessitate the use of models that are robust and reliable over a broad range of spatial domains and time scales. To achieve this goal, a multi-faceted theoretical approach is taken that combines quantum chemistry, molecular simulation and statistical mechanical methods, many of which have been the development focus of the proposed research group of Prof. Darrin York over the course of the last several years. These methods are collectively referred to as ''multi-scale'' models for RNA catalysis. In this context, the two main milestones in this project of postdoctoral research involve the development, calibration and validation of QM/MM models for RNA and other phosphoryl transfer reactions, and the extension of fast, linear-scaling electrostatic and solvation methods for QM/MM calculations with more general ab initio charge distributions and polarizable force fields. The application focus is on important phosphoryl transfer reactions in solution for which there are abundant recent experimental measurements of rate profiles, linear free-energy relations and kinetic isotope effects.
Computational Analysis of Organometallic Reactions
The Stille Reaction
The impact that metal-catalyzed cross-coupling processes, in particular the Stille and the Suzuki reactions, have on synthesis is unquestionable. High selectivity, functional group tolerance and mildness of the reaction conditions make these processes valuable for C-C bond formation even in the complex setting of natural product synthesis. However, the choice of reaction conditions (i.e., palladium source, additives, solvents, halide or pseudohalide, ligands, the "copper co-catalysis", etc.) is still rather empirical.Accountig for those variables, multifaceted reaction mechanisms (more comprehensive than the classical oxidative addition, transmetalation and reductive elimination) are emerging. Casado and Espinet proposed two associative mechanisms for the transmetalation step to account for the reaction rates in the coupling of aryltriflates and tributil vinyl stannane, which accounted for the stereochemical outcome in the coupling of some chiral non-racemic alkyl stannanes. It was found that, for the coupling of X-bridging electrophiles and stannanes, the associative mechanism that proceeds through a cyclic transition state (or intermediate) resulting in the replacement of a ligand by the stannane transfering substituent, was favored over the alternative open transition state.
On model systems (vinylbromide and tributylvinyltin with Pd(PMe3)2) we have characterized the associative transmetalation mechanism and a cyclic 4-atom transition state after ligand dissociation, thus giving credence to Espinet proposal and justifying the retention of configuration when using chiral non-racemic alkyl stannanes as coupling partners.
The gold(I)-catalyzed Rautenstrauch rearrangement
The synthesis of cyclopentenones from alkynyl allyl acetates under catalysis by Pd (Rautenstrauch rearrangement) has been revisited by Toste using Gold(I) salts to promote the reaction. Our computational studies on the mechanism of the rearrangement, in particular the center-to-helix-to center chirality transfer, indicate that they depart from the classical Pd mechanism, and ocurr by electrophilic activation of the alkyne, transfer of the carboxyl group to the Csp atom and cyclization of the gold-pentadienyl intermediate without carboxylate rotation or helix interconversion, thus ensuring fidelity in the chirality transfer process.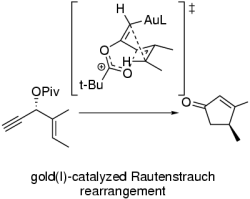
J. Am. Chem. Soc. 2006, 128, 24 34-2437
Computational Analysis of Pericyclic and Other Concerted Reactions
Lemal et al. described in 1976 that certain extremely facile automerization processes ocurring at low temperature appeared to ignore the Woodward-Hoffmann rules, and proposed the existence of a new subset of reactions termed pseudopericyclic.From a theoretical point of view, the distinction between pericyclic and pseudopericyclic reactions is not straightforward. In previous theoretical studies of these processes, Birney concluded that prototypical pseudopericyclic reactions exhibit three main features:
- they can be orbital symmetry allowed regardless of the number of electrons involved
- the energy barriers can be very low or even non-existing
- they have a planar transition state, if possible.
Pericyclic reactions of charged species (carbocations, carbanions) are currently being computationally addressed. Systems of interest are:
The solvolytic ring-opening of cyclopropylhalides and the Woodward-Hoffmann-DePuy rule
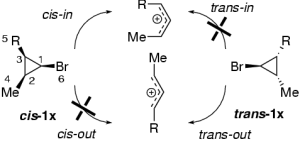
J. Org. Chem. 2004, 69, 9002-9010
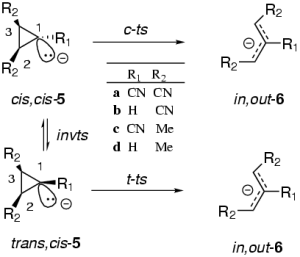
The ring-opening of cyclopropyl anions
The ring-closure of hydroxypentadienyl cations (Nazarov and the like)
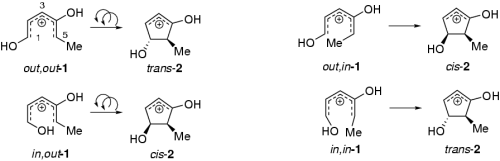
Chem. Eur. J. 2004, 10, 4324-4333
Electrocyclic, sigmatropic and cheletropic reactions of (di)vinylallenes and derivatives (vinyallene oxides, cyclopropyl analogs..)
J. Org. Chem. 2004, 69, 3635-3644
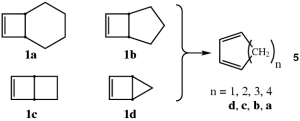
Electrocyclic reactions of bicyclic systems in a quest for anti-Woodward-Hoffmann behavior
Cyclopentannelation of allenolate allyl carbamates
In the frontier between concerted and stepwise processes, we have computed the mechanism of the chirality transfer in the cyclopentannelation of allenolate allyl carbamates, showing a subtle mechanistic duality with features of both pericyclic and ionic processes.